Sickle Cell Disease
Sickle cell disease is an inherited blood disorder that affects red blood cells. It is a type of hereditary haemoglobin disorder where valine has been substituted for glutamic acid at position 6 of haem beta chain, caused by a point mutation. You must remember that Sickle Cell Disease (SCD) is inherited in a Mendelian recessive manner. Therefore patients with two Sickle genes (SS) or carry one S gene but with concomitant Beta Thalassemia ( SB) are affected.

This disease is common in peoples of Equatorial African ancestry.
Clinical Presentations
Remember that all clinical features are due to two main features of the disease- haemolysis and vaso-occlusive crisis. You must understand that HbS is insoluble in the dexoygenised form and they have a shorter life span due to increased fragility, therefore causing chronic haemolysis ( similar to Thalassemia). The red blood cells with HbS also tend to aggregrate and cause thrombosis, this will leads to tissue infarction. Remember that the vaso-occlusive crisis tends to be precipitated by HADI ( hypoxia, acidosis,dehydration and infection)
Clinical features due to haemolysis
Anemia
Gallstone
Bone marrow enlargement
( these features also occur in Thalassemia patients)
Clinical fatures due to vaso-occlusive crisis
Bone pain- may cause vascular necrosis
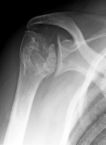 Humerol head avscular necrosis
Humerol head avscular necrosis
Leg ulcers
Genito-urinary- priapism
Cerebral-stroke
Spleen- initially may cause splenomegaly due to extra medullary haemopoiesis ( due to anemia) but later splenic infarct and hyposplenism. Patients tend to have capsulated bacteria infection and Salmonella osteomyelitis.
Chest-acute chest pain
( remember that these are all due to thrombotic events)
Physical Signs
Patients tend to be pale with jaundice. Hepatomegaly may be present. Look for chronic leg ulcers.
Investigations
Full blood count- low Hb with features suggesting haemolysis such as increased reticulocyte counts
LFT- increased bilirubin and AST
Peripheral blood film- sickled cells
Haemoglobin electrophoresis- to determine variant haemoglobin
X-ray- to look for vascular necrosis if patients present with joint pain.
Management
During sickle crisis ( oxygen, analgesia, rehydration)
Long term management
- prophylactic penicillin for prevention of penumococcal infection,
- management of anemia, however be careful about secondary iron overload due to multiple transfusions
- folate supplements
Tips for MRCP
1) Always suspect Sickle Cell Disease if a patient has anemia and chronic leg ulcers.

